Diagnostica differenziale delle ipereosinofilie.
Caso clinico
Maschio, 68 anni, macellaio, ex fumatore dalla giovane età fino a 50 anni. Abitudini di vita regolari, segnala di consumare carne cruda bovina e suina circa 1 volta alla settimana. Alvo raramente diarroico in occasioni isolate, diuresi regolare, non riferisce allergie.
In anamnesi a 20 anni riferisce pleuro-polmonite da lato imprecisato, insorta durante il servizio militare. Iperteso da 10 anni. 7 anni fa fu coinvolto in un incidente stradale con frattura trocanterica femorale trattata con osteosintesi. Successivamente vennero rimossi i mezzi di osteosintesi.
Noto per ostruzione nasale cronica da ipertrofia polipoide dei turbinati studiata con test allergologici risultati negativi. In passato aveva rifiutato la risoluzione chirurgica.
2 anni fa fu curato per la frattura traumatica del piatto tibiale destro e sottoposto ad ernioplastica inguinale destra.
Circa 1 anno fa riferisce bronchite a lenta risoluzione.
Ad agosto del corrente anno venne posta la diagnosi clinica di broncopolmonite, per questo aveva assunto levofloxacina. In tale occasione i controlli ematici dimostravano la presenza di leucocitosi (GB 13.000 mcl. EO 29 %). Dopo un apparente miglioramento ricomparve febbre, non preceduta da brivido, tosse associata a sibili, dispnea da sforzo e peggioramento della nota ostruzione nasale. La radiografia del torace mostrava un addensamento di tipo interstizio-alveolare ed un aumento diffuso del disegno polmonare. Gli esami ematici rilevavano: GB 26.000 Eosinofili 55 %, VES 60, PCR 4.6 (vn.
Obiettivamente era presente un’importante ostruzione al flusso aereo delle coane nasali con rinolalia, il murmure vescicolare risultava ridotto ed erano presenti gemiti e sibili soprattutto a carattere espiratorio ed espirio prolungato. Negativo il restante esame obiettivo.
All’ingresso veniva registrato un ECG a riposo che mostrava la presenza di una transizione anticipata dell’onda R e T negative in sede anteriore estesa; dosata subito la troponina, essa risultava positiva (0.61, vn. < 0.10 ng/ml). Venne discusso il caso con il Cardiologo ed eseguita un’ecocardiografia che rilevava un ventricolo sinistro di dimensioni conservate con un’ipocinesia diffusa e frazione di eiezione ridotta al 40 %. Era presente ipertrofia del setto interventricolare ed un aspetto rifrangente del miocardio ed evidenza di ispessimento dei lembi della valvola mitrale. Le sezioni destre erano normali, era presente un minimo versamento pericardico di 6 mm.
Eco addome: fegato lievemente ingrandito con ecostruttura finemente addensata. Nella parete della colecisti erano presenti polipi colesterinici. Il restante reperto era nei limiti della norma, in particolare la milza aveva il diametro maggiore di 10.5 cm e quello minore di 4.8 cm.
Es ematici: GB 27.450, neutrofili 19 %, linfociti 12 %, monociti 2.7 %, eosinofili 65 %, VES 88, PCR 2.6. Tutti gli altri esami di routine nella norma, all’elettroforesi delle sieroproteine era presente un aumento delle γglobuline pari al 29.4 % (22 gr %), riduzione dell’albumina al 42 % (3.24 gr %). Aumento delle α2-globuline al 13 % (1 gr %). ANA, ENA, ANCA, marker tumorali comprese enolasi acide e cromogranina, negativi. ACE, ANA, ANCA, anticorpi anti-gliadina, -endomisio, -transglutaminasi, β2-microglobulina: negativi. Esami parassitologici delle feci negativi. Incremento delle IgE a 4599 (vn.
Per inquadrare la sindrome ipereosinofila vennero eseguiti anche esofago-gastro-duodenoscopia e colonscopia. La gastroscopia mostrò la presenza di erosioni che vennero sottoposte ad esame bioptico, dal quale emerse un quadro di gastrite cronica aspecifica. Il Bulbo duodenale si presentava con mucosa edematosa ma privo di lesioni, la biopsia eseguita mostrava lieve flogosi cronica. Alla colonscopia il reperto endoscopico risultava normale e il materiale prelevato, compreso quello ottenuto dall’ultima ansa ileale, risultò indicativo per un quadro di lieve flogosi cronica aspecifica.
Venne sottoposto a biopsia anche un polipo nasale, l’istologia del quale mostrò, anche in questo caso, una flogosi aspecifica senza una particolare infiltrazione eosinofila.
Venne eseguito lo studio molecolare degli eosinofili dal sangue periferico per escludere proliferazioni maligne degli stessi: ricerca mutazione BCR/ABL (traslocazione 9-22) e 5-12; 8-22, mutazione FIP1/L1/PDGFR ALFA + WT1. Tutti risultarono negativi. L’immunofenotipo del sangue periferico non ha rilevato disordini clonali della popolazione T.
Considerato l’interessamento miocardico e polmonare nonché lo stato di sofferenza respiratoria del paziente si iniziò un trattamento con una piccola dose di steroide: Prednisone 25 mg/die (circa 0.35 mg/kg). In virtù del ritrovamento di onde T negative e di rialzo della troponina. Vennero associati aspirina 100 mg ed eparina a basso peso molecolare a dosi anticoagulanti. Iniziato 1.25 mg di bisoprololo e ramipril 5mg. Venne iniziata protezione gastrica con inibitori della pompa protonica contemporaneamente ad una associazione di steroide e β2-agonista adrenergico long acting per via inalatoria, con l’aggiunta di tiotropio sempre per via inalatoria.
Contemporaneamente venne discusso il caso con l’Infettivologo, con l’intento di escludere un’infezione parassitaria occulta e venne stabilito di associare un trattamento antielmintico ad ampio spettro con mebendazolo. Terminato il primo ciclo, ne venne effettuato un secondo a 15 giorni di distanza. Contemporaneamente la moglie riferiva ipersonnia e roncopatia con apnee notturne. Venne per questo eseguita una registrazione della saturimetria notturna che dimostrava un tracciato compatibile con la presenza di apnee ostruttive che vennero risolte con una piccola dose inalatoria di ossigeno notturno (1 litro al minuto). Il problema verrà approfondito dopo la stabilizzazione della malattia eosinofila.
Dopo circa 7 giorni di terapia steroidea gli addensamenti polmonari regredirono, con normalizzazione delle onde T negative, rilevate all’ECG e della troponina. Il Paziente venne dimesso in decima giornata clinicamente guarito ma con la raccomandazione di proseguire la terapia steroidea e il follow-up clinico-laboratoristico del caso. La terapia steroidea venne programmata con una riduzione progressiva, ma lenta, della dose giornaliera, tenendo controllato l’esame obiettivo e l’emocromo . Attualmente gli eosinofili sono normalizzati e la clinica è negativa.
Discussione
I disordini mieloproliferativi comprendono 1) la leucemia mieloide cronica, caratterizzata dalla presenza del gene di fusione BCR-ABL, più comunemente risultante dalla traslocazione 9:22 (Cromosoma Philadelphia); 2) la policitemia vera; 3) la trombocitemia essenziale; 4) la mielofibrosi idiopatica. Condizioni meno comuni che vengono però incluse dall’organizzazione mondiale della sanità in questo capitolo sono la mastocitosi, la leucemia mielomonocitica cronica, la leucemia neutrofila cronica, e la leucemia eosinofila cronica (6).
La sindrome ipereosinofila invece si definisce come un’ipereosinofilia (eosinofili maggiori o uguali a 1500 per microlitro = mcl) persistente da almeno 6 mesi nel sangue periferico, in assenza di cause secondarie (7) (vedere Tabella 1, 8). E’ caratteristica del sesso maschile, nella maggioranza dei casi fra 20 e 50 anni(9-10-11). Infrequentemente è stata dimostrata un’alterazione cariotipica clonale e in questo caso si parla di leucemia eosinofila cronica (4), inserita come detto sopra, fra i disordini mieloproliferativi.
In letteratura si distinguono l’eosinofilia dall’ipereosinofilia poiché nel primo caso gli eosinofili sono maggiori di 500, ma inferiori a 1500, nella seconda sono superiori a 1500 mcl, altri distinguono un’eosinofilia lieve fra 351 e 1500 eosinofili per mm3, moderata maggiore di 1500 fino a 5000 eosinofili per mcl maggiore di 5000 eosinofili per mm3 (12)(1, figura 3).
Se la diagnostica differenziale verso le ipereosinofilie secondarie risultasse negativa allora si dovrà procedere allo studio degli eosinofili periferici e, nel caso dovessero essere rilevarti dei marcatori di clonalità, si renderebbe necessaria l’esecuzione di un aspirato e di una biopsia osteomidollare (13-14-15).(Tabella N.1, figura 4)
Le cause più comuni di eosinofilia sono quelle infettive, soprattutto da infestazione elmintica. Se il Paziente non è stato in luoghi a rischio ed è a contatto con cani è probabile che sia in causa la Toxocara Canis, verme rotondo trasmesso per via orofecale, per la cui diagnosi va eseguita la sierologia. Se è stata consumata carne di maiale mal cotta è possibile sia in causa la Trichinella Spiralis un altro verme rotondo: per porre la diagnosi è necessaria la biopsia muscolare. Importante è escludere anche l’infestazione da Strongiloides Stercoralis, verme rotondo della grandezza di pochi mm che può infestare i Pazienti sottoposti a chemioterapia e/o blocco dell’acidità gastrica per lungo tempo con inibitori della pompa protonica che evitano così l’abbattimento della flora batterica nel bolo alimentare. Va eseguita anche la sierologia per la Toxoplasmosi e se vi è il dubbio di esposizione anche per lo Schistosoma. In quest’ultimo caso è necessario eseguire anche una ricerca nelle urine e una biopsia della vescica. La seconda causa più frequente di ipereosinofilia è la presenza di allergia o atopia (16-17). Possono provocare eosinofilia anche il lupus eritematoso sistemico, l’artrite reumatoide, la sindrome di Churg-strauss (vasculite sistemica con asma), la panarterite nodosa e rare malattie come quella di Kimura, flogosi cronica idiopatica che si manifesta con tumefazioni sottocutanee non dolorose nella regione del capo, del collo e/o delle ghiandole salivari; la malattia di Wells, dermatite granulomatosa ricorrente; la malattia di Castelman anche chiamata iperplasia angiofollicolare dei linfonodi; o una malattia infiammatoria intestinale. Vanno escluse anche la sindrome di Job (18), il deficit di IgA, e la sindrome di Wiskott-Aldrich (è un disordine congenito descritto per la prima volta nel 1937 da Wiskott, attualmente ne sono descritti circa 300 casi, si presenta con emorragia da piastrinopenia con micropiastrine, eczema e frequenti infezioni. Nella maggioranza dei pazienti sono presenti difetti a carico sia dei linfociti T che di quelli B) con il dosaggio delle immunoglobuline (19-20). Farmaci come fenitoina, carbamazepina, fenobarbital, fra gli anticonvulsivanti, ranitidina, antibiotici sulfamidici, l’allopurinolo, gli stimolatori delle colonie dei granulociti e dei macrofagi possono provocare ipereosinofilia e sintomi sistemici. La sindrome eosinofilia-mialgia è correlata all’assunzione di L-triptofano presente in quantità diversa negli integratori distribuiti specie alle persone dedite al culturismo o all’ingestione di cibo cucinato con oli contenenti anilina.
Dal punto di vista pneumologico si deve escludere: la fibrosi cistica, l’aspergillosi broncopolmonare allergica che può complicare il decorso di un’asma, e l’infiltrato polmonare di Loeffler (21). Quest’ultimo provoca degli infiltrati eosinofili fluttuanti. Vanno esclusi anche problemi gastroenterologici come la celiachia e la gastroenterite eosinofila.
Fra le cause endocrine di ipereosinofilia va presa in considerazione l’insufficienza surrenale.
Fra le cause ereditarie è nota l’eosinofilia familiare. Questa è ereditata con una modalità autosomica dominante. Il nostro Paziente non presenta una storia familiare di eosinofilia né la tipica facies (cute ruvida, asimmetria facciale, fronte prominente, occhi infossati, radice nasale larga e punta del naso carnosa, prognatismo) (22-23-24).
Le neoplasie che provocano eosinofilia sono molteplici: le malattie linfoproliferative e i carcinomi, specialmente quelli di rene, polmone e mammella. Molte malattie ematologiche provocano eosinofilia per produzione di IL-3 e 5 per esempio il linfoma a cellule T, il linfoma di Hodgkin e la leucemia linfoblastica acuta. Alcune neoplasie ematologiche associate ad eosinofilia colpiscono soprattutto l’età pediatrica (9-10-11). La leucemia linfoblastica acuta, nella maggior parte dei casi è un clone di cellule precursori della linea B. La leucemia mieloide acuta con un’inversione pericentrica del cromosoma 16 presenta un’eosinofilia midollare ma solitamente non periferica (25-26). Recentemente è stata descritta un’eosinofilia midollare e periferica associate ad un linfoma linfoblastico costituito da precursori di cellule-T associato ad una traslocazione tra il cromosoma 8 e il 13 (27).
Gli eosinofili contengono granulazioni molto tossiche se rilasciate in quantità eccessiva nei tessuti come accade nell’ipereosinofilia. Per questo vari tessuti dell’organismo possono essere danneggiati: la cute, il sistema nervoso centrale, i visceri cavi e parenchimatosi. Il riscontro nell’aspirato midollare osseo di eosinofili ipogranulati con corpi lipidici è indice di attivazione di queste cellule (28).
Le mutazioni geniche sopra riportate e caratteristiche di alcune forme di ipereosinofilia sono: la traslocazione t(1;4)(q44;q12) (tavola A) è diagnostica di un oncogene legato alla malattia. Questa traslocazione è stata trovata non in tutti i casi di sindrome ipereosinofila ma in quelle che vengono, in base a questo riscontro, definite come leucemie eosinofile croniche. La lesione principale è quella che si evidenzia nella tavola B che provoca la fusione di due geni del braccio lungo del cromosoma 4: il PDGFRA e il FIP1L1. Questo provoca la sintesi di una tirosinchinasi attiva (29).
La produzione di eosinofili richiede 3 citochine: interleukina (IL) -5, IL-3, e il fattore di crescita delle colonie di granulociti-macrofagi (GM-CSF). In rari casi delle cellule linfomatose o leucemiche sovraproducono IL-5 simulando un’ipereosinofilia primitiva(29). Nella maggior parte dei casi di sindrome ipereosinofila idiopatica gli eosinofili sono indipendenti dai fattori di crescita e le caratteristiche cliniche sono quelle di una malattia mieloproliferativa con epatosplenomegalia e trombocitemia. In altri casi di sindrome ipereosinofila una popolazione clonale di T-cellule si mescola assieme agli eosinofili. Queste T cellule clonali sono attivate e mostrano dei riarrangiamenti anormali dei marker di superficie (30). Queste T-cellule producono abbondanti quantità di IL-5 che presumibilmente causa l’ipereosinofilia. Le IgE in circolo possono essere molto elevate in conseguenza di altre citochine rilasciate sempre dall’anzidetta popolazione T. cellulare. Molti Pazienti con questa variante presentano una dermatite intrattabile e alla biopsia cutanea presentano un infiltrato T suggestivo di un linfoma cutaneo T (31-32-33).
La sindrome ipereosinofila idiopatica provoca sintomi costituzionali, come nel nostro Paziente, nel 50 % dei casi (astenia, affaticabilità, anoressia, febbre, calo ponderale, mialgie); in più del 70 % sintomi cardiopolmonari (tosse, dispnea, insufficienza cardiaca, aritmie, malattia dell’endo-miocardio, infiltrati polmonari, versamento pleurico, embolia)(34). In più del 50 % causa problemi ematologici (tromboembolie, anemie, trombocitopenie, adenopatie, splenomegalia)(35-36). Le alterazioni neurologiche compaiono in più del 50 % (alterazioni del comportamento e delle funzioni intellettuali, spasticità, neuropatie periferiche, lesioni cerebrali focali)(37-38). Lesioni dermatologiche compaiono in oltre il 50 % (dermografismo, angioedema, eruzioni, prurito, ascessi “freddi”)(39-40-41-42). In più del 40 % i sintomi sono gastrointestinali (diarrea, nausea, crampi addominali). Tra le alterazioni ematologiche che compaiono in circa il 40 % dei Pazienti vi sono l’incremento delle immunoglobuline specialmente IgE e degli immunocomplessi circolanti. (Vedere tabella 1)
Il trattamento d’elezione in questi Pazienti, che sono negativi per le mutazioni geniche sopra riportate, rimane il Prednisone, seguito dall’Idrossiurea e dall’interferon alfa (28-43). Nei casi refrattari si usa il clorambucil, l’etoposide e la vincristina (44-45-46-47).
Nei casi invece in cui è presente una mutazione del PDGFRA e il FIP1L1 la terapia più moderna prevede l’uso in prima istanza dell’Imatinib (48).
Recentemente nei casi refrattari soprattutto in quelli in cui sono stati riconosciuti disordini linfoproliferativi clonali delle cellule T, associati spesso a lesioni cutanee si è riscontrato che l’interleukina 5 ha un ruolo critico. Per questo sono stati usati due anticorpi monoclonali contro l’Interleukina 5 (IL 5): l’alemtuzumab e il mepolizumab (49-50-51), utile nei casi refrattari anche l’anticorpo monoclonale contro l’antigene CD 52 precedentemente menzionato (52-53). L`antigene CD52 viene espresso con elevata densità da linfociti, monociti, eosinofili, timociti e macrofagi. Questo antigene viene espresso dalla maggior parte dei tumori maligni di origine linfoide, sebbene l`espressione sulle cellule di mieloma sia variabile
Nei casi refrattari e in cui è possibile rimane il trapianto allogenico di midollo osseo (54-55-56-57).
Conclusioni:
La sindrome ipereosinofila idiopatica è una malattia cronica che si manifesta in modo polimorfo: con sindromi cardiopolmonari, ematologiche, neurologiche, dermatologiche, gastrointestinali o costituzionali come in questo caso.
E’ necessario seguire con attenzione negli anni il Paziente in modo da sincerarsi che non si manifestino nel tempo altre patologie che renderebbero non più “Idiopatica” l’ipereosinofilia.
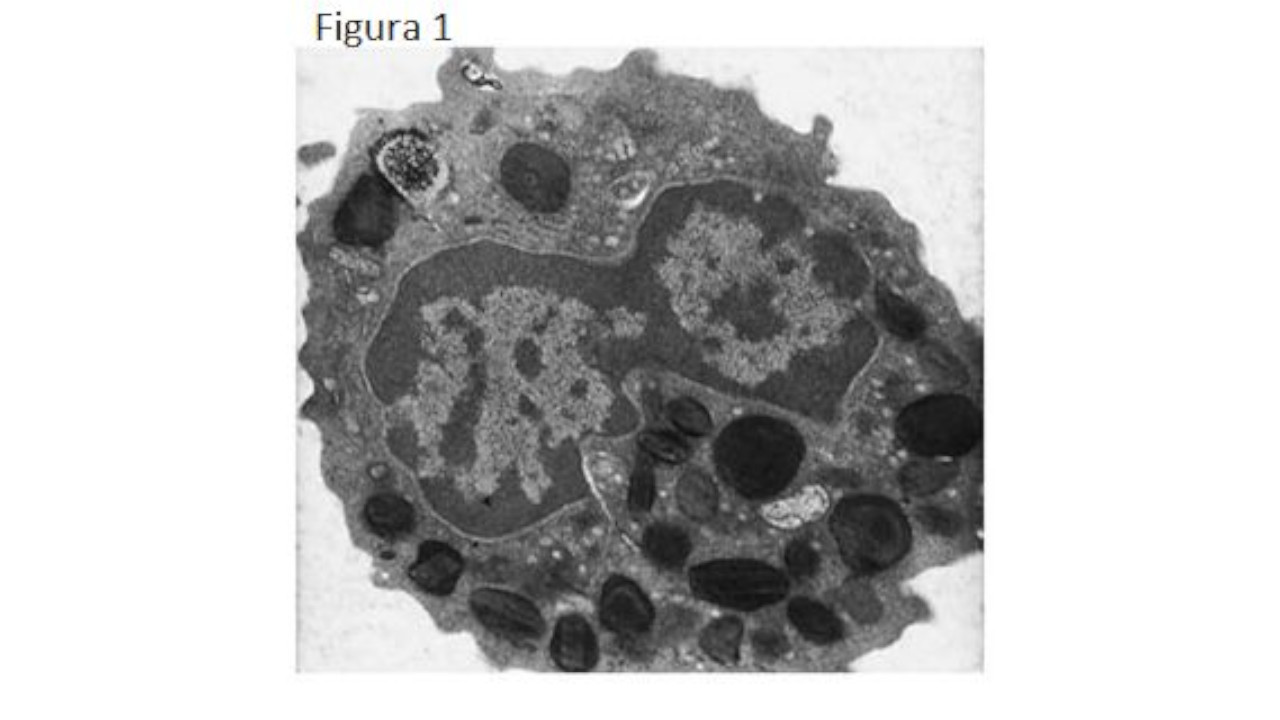
Un eosinofilo al microscopio elettronico.
Il nucleo a forma di “pince-nez” (occhiali a molla) circondato da granuli, molti dei quali anno all’interno una formazione centrale elettrondensa. Questa formazione è una proteina basica tossica. La matrice che circonda la formazione centrale del granulo contiene proteine tossiche cationiche e perossidasi. (Schwartz 348 (13): 1199, Figure 1 March 27, 2003)
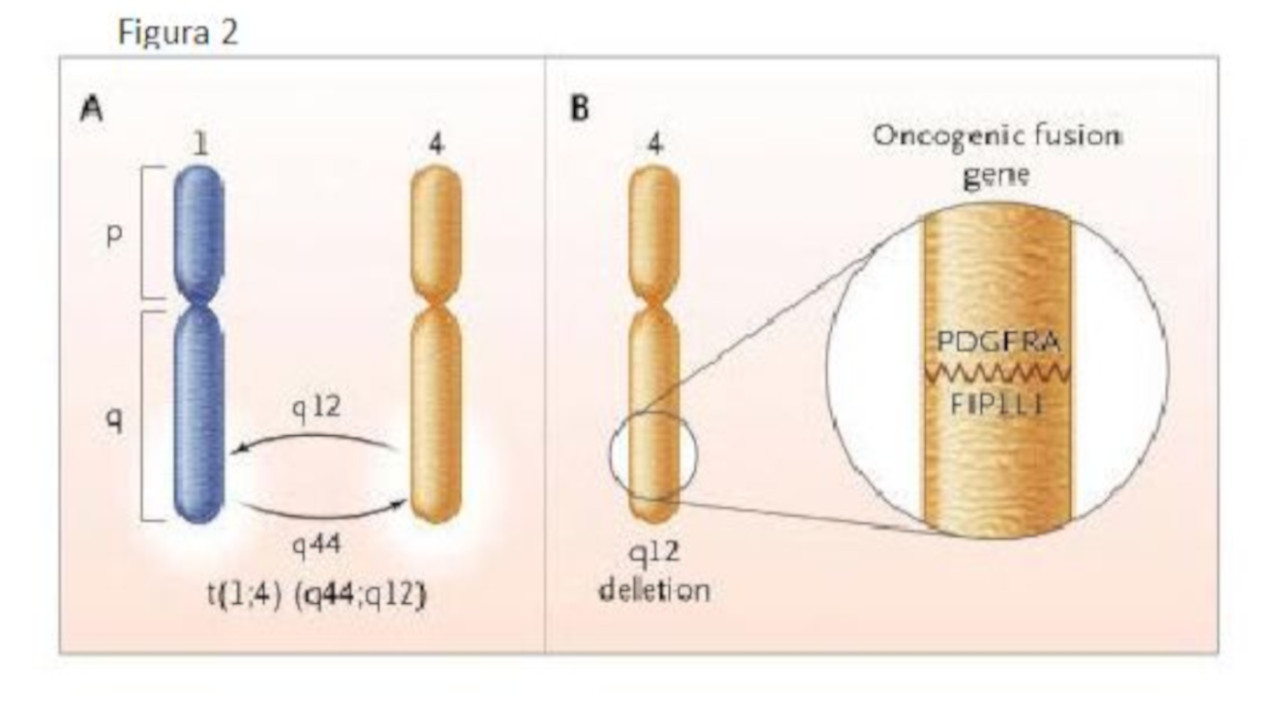
Le alterazioni cromosomiche nella sindrome ipereosinofila.
La traslocazione t(1;4)(q44;q12) translocation (tavola A) è diagnostica di oncogene legato alla malattia. Questa traslocazione è stata trovata non in tutti i casi di sindrome ipereosinofila ma in quelle che vengono, in base a questo riscontro, definite come leucemie eosinofile croniche. La lesione principale è quella che si evidenzia nella tavola B che provoca la fusione di due geni del braccio lungo del cromosoma 4: il PDGFRA e il FIP1L1. Questo provoca la sintesi di una tirosinchinasi attiva.
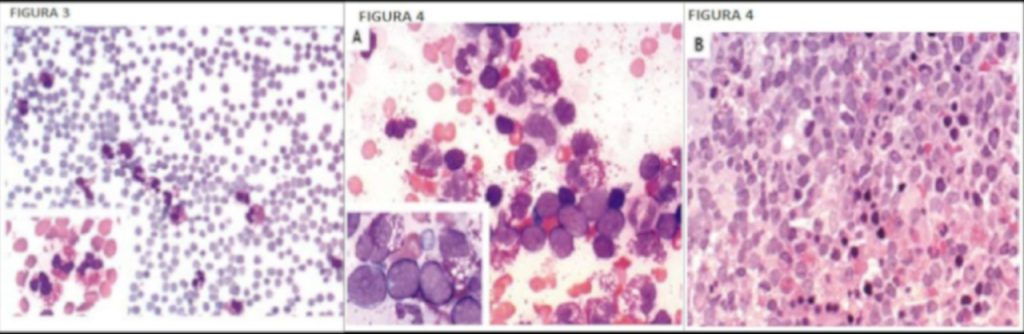
Figura 3 (2). (Figura in fondo alla pagina)
Striscio periferico
Leucocitosi con marcata eosinofilia. Gli eosinofili includono molte forme anormali con nuclei trilobati e granulazioni citoplasmatiche. (Wright–Giemsa, x200; riquadro, x1000)
Figura 4. (Figura in fondo alla pagina)
Aspirato midollare e biopsia ossea.
L’aprirato midollare (Figura A) contiene prevalentemente una popolazione di eosinofili mature e blasti con citoplasma ridotto e agranulare. (Wright–Giemsa, x500; riquadro, x1000). Il campione bioptico (Figura B) è marcatamente ipercellulare, con eosinofili maturi e elementi eritroidi.
Tabella N. 1 (2)
| Cause di eosinofilia | ||||||
| Malattie infettive | Infezioni parassitarie | Infezioni fungine | ||||
| Disordini immunologici | Allergia | Disordini autoimmuni | Malattie infiammatorie intestinali | |||
| Tossine | Ambientali | Farmaci | ||||
| Cause endocrine e metaboliche | ||||||
| Cause genetiche e congenite | ||||||
| Sindromi ipereosinofile | Idiopatiche | altre | ||||
| Cause neoplastiche | Non ematopoietiche | Carcinoma del piccolo intestino | Carcinoma dell’ovaio | Carcinoma del polmone | Carcinoma del pancreas | Altri organi solidi |
| Ematopoietiche | Linfoma di Hodgkin | Leucemia eosinofila | Mastocitosi | |||
Figura 5. Processi coinvolti nell’ipereosinofilia (1)
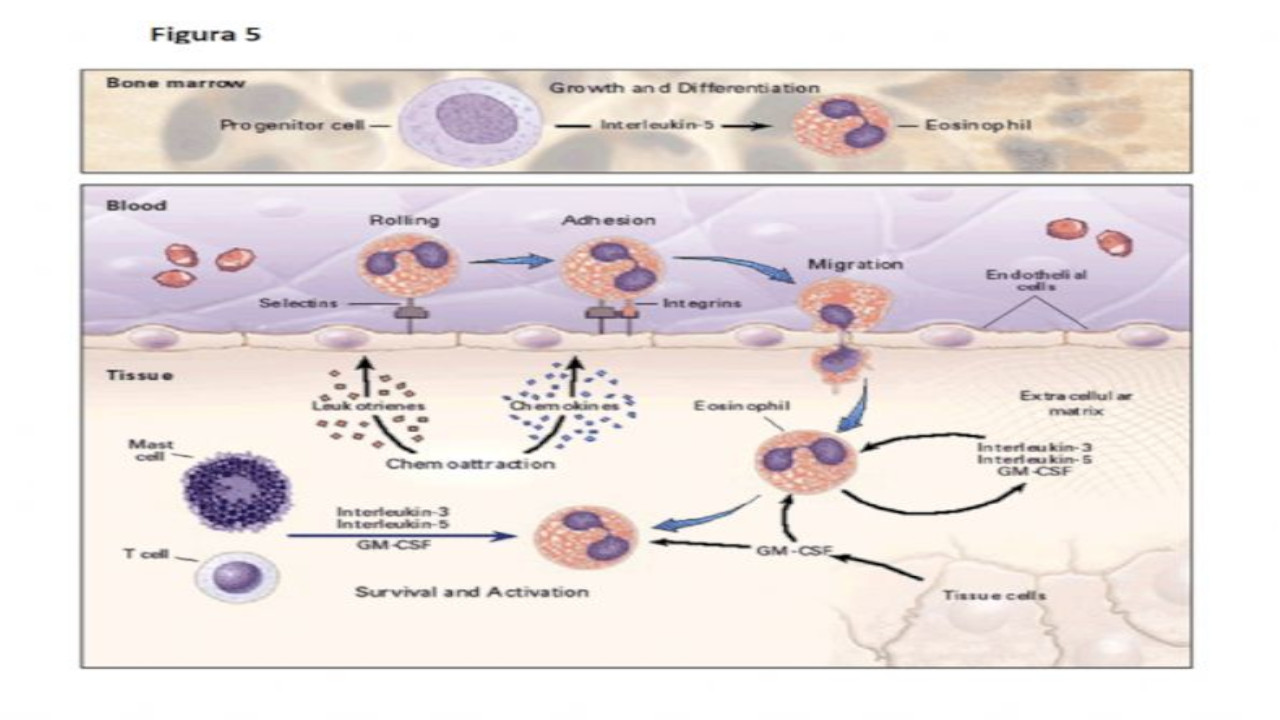
Gli eosinofili si sviluppano nel midollo osseo in risposta alla stimolazione delle cellule progenitrici da parte della interleukina-5. Gli eosinofili maturi aderiscono alle cellule endoteliali attraverso l’interreazione delle selettine e delle integrine con i loro rispettivi recettori endoteliali. Dopo l’esposizione a mediatori chemoattraenti, gli eosinofili attraversano l’endotelio per diapedesi e migrano nei tessuti. L’accumulo di eosinofili è regolato dalla produzione di fattori da parte di cellule T e probabilmente di mastcellule che aumentano la loro sopravvivenza e li attivano (IL-3, IL-5, fattore stimolante le colonie granulocitarie e macrofagiche. Gli esosinofili stessi sono capaci di generare le citochine che prolungano la loro sopravvivenza in risposta a componenti della matrice extracellulare.
BIBLIOGRAFIA
1) M. E. Rothenberg. Eosinophilia. NEJM 1998; 338: 1592-1600
2) Huang and Hasserjian. NEJM; 350 (25): 2604; 2004
3) Robert S.Schwartz. The Hypereosinophilic Syndrome and the biology of cancer. NEJM; 348 (13): 1199-1200; 2003
4) E. Messa, D. Cilloni, G. Saglio. A young man with persistent eosinophilia. Inter Emerg Med 2: 107-112, 2007
5) Sabine-Gisela Plotz, Hanss-Uwe Simon, Ulf Darsow et altri. Use of Anti-Interleukin-5 Antibody in the Hypereosinophilic Syndrome with Eosinophilic Dermatitis. NEJM; 349:2334-2339; 2003
6) Peter J Campbell,AntonyR. Green. The Myeloproliferative Disorders. NEJM; 355:2452-2466; 2006
7) Chusid MJ, Dale DC, West BC, Wolff SM. The hypereosinophilic syndrome: analysis of fourteen cases with review of the literature. Medicine (Baltimore) 1975; 54:1.
8) Harrison. Principles of Internal Medicine. XVIII edizione.
9) Weller PF, Bubley GJ. The idiopathic hypereosinophilic syndrome. Blood 1994; 83:2759.
10) Farruggia P, D`Angelo P, Acquaviva A, et al. Hypereosinophilic syndrome in childhood: clinical and molecular features of two cases. Pediatr Hematol Oncol 2009; 26:129.
11) Rapanotti MC, Caruso R, Ammatuna E, et al. Molecular characterization of paediatric idiopathic hypereosinophilia. Br J Haematol 2010; 151:440.
12) Valent P, Klion AD, Horny HP, et al. Contemporary consensus proposal on criteria and classification of eosinophilic disorders and related syndromes. J Allergy Clin Immunol 2012; 130:607.
13) Swerdlow SH, Campo E, Harris NL, et al. World Health Organization classification of tumours of haematopoietic and lymphoid tissues, IARC Press, Lyon 2008.
14) Bain BJ. Relationship between idiopathic hypereosinophilic syndrome, eosinophilic leukemia, and systemic mastocytosis. Am J Hematol 2004; 77:82.
15) Fridman A, Sagiv Sh, Shvidel L, Berrebi A. Reversible Horner syndrome caused by solitary plasmacytoma of second thoracic vertebrae. Leuk Lymphoma 2004; 45:2531.
16) Shyur SD, Hill HR. Immunodeficiency in the 1990s. Pediatr Infect Dis J 1991; 10:595.
17) Buckley RH. Other well-defined immunodeficiency syndromes (The Hyper-IgE Syndrome). In: Immunologic disorders in infants and children, 5th ed, Stiehm ER, Ochs HD, Winkelstein JA (Eds), Elsevier Saunders, Philadelphia 2004. p.550.
18) Davis SD, Schaller J, Wedgwood RJ. Job`s Syndrome. Recurrent, “cold”, staphylococcal abscesses. Lancet 1966; 1:1013.
19) Pien GC, Orange JS. Evaluation and clinical interpretation of hypergammaglobulinemia E: differentiating atopy from immunodeficiency. Ann Allergy Asthma Immunol 2008; 100:392.
20) Ozcan E, Notarangelo LD, Geha RS. Primary immune deficiencies with aberrant IgE production. J Allergy Clin Immunol 2008; 122:1054.
21) Fauci AS, Harley JB, Roberts WC, et al. NIH conference. The idiopathic hypereosinophilic syndrome. Clinical, pathophysiologic, and therapeutic considerations. Ann Intern Med 1982; 97:78.
22) Minegishi Y, Saito M, Tsuchiya S, et al. Dominant-negative mutations in the DNA-binding domain of STAT3 cause hyper-IgE syndrome. Nature 2007; 448:1058.
23) Gene Reviews. https://www.ncbi.nlm.nih.gov/books/NBK25507 (Accessed on June 02, 2010).
24) Minegishi Y, Saito M, Morio T, et al. Human tyrosine kinase 2 deficiency reveals its requisite roles in multiple cytokine signals involved in innate and acquired immunity. Immunity 2006; 25:745.
25) Spry CJF. Idiopathic hypereosinophilic syndrome. In: Eosinophils: Biological and clinical aspects, Makino S, Fukuda T (Eds), CRC, Boca Raton 1992. p.403.
26) Ackerman SJ, Bochner BS. Mechanisms of eosinophilia in the pathogenesis of hypereosinophilic disorders. Immunol Allergy Clin North Am 2007; 27:357.
27) Roufosse F, Cogan E, Goldman M. Lymphocytic variant hypereosinophilic syndromes. Immunol Allergy Clin North Am 2007; 27:389.
28) Ogbogu PU, Bochner BS, Butterfield JH, et al. Hypereosinophilic syndrome: a multicenter, retrospective analysis of clinical characteristics and response to therapy. J Allergy Clin Immunol 2009; 124:1319.
29) Bain BJ, Fletcher SH. Chronic eosinophilic leukemias and the myeloproliferative variant of the hypereosinophilic syndrome. Immunol Allergy Clin North Am 2007; 27:377.
30) Klion AD, Noel P, Akin C, et al. Elevated serum tryptase levels identify a subset of patients with a myeloproliferative variant of idiopathic hypereosinophilic syndrome associated with tissue fibrosis, poor prognosis, and imatinib responsiveness. Blood 2003; 101:4660.
31) Buckley RH, Wray BB, Belmaker EZ. Extreme hyperimmunoglobulinemia E and undue susceptibility to infection. Pediatrics 1972; 49:59.
32) Hill HR, Quie PG. Raised serum-IgE levels and defective neutrophil chemotaxis in three children with eczema and recurrent bacterial infections. Lancet 1974; 1:183.
33) Cho C, Ferdman RM, Church JA, Ong PY. Skin-deep clues to a complex disease. Ann Allergy Asthma Immunol 2010; 104:93.
34) Paulson ML, Freeman AF, Holland SM. Hyper IgE syndrome: an update on clinical aspects and the role of signal transducer and activator of transcription 3. Curr Opin Allergy Clin Immunol 2008; 8:527.
35) Wang JG, Mahmud SA, Thompson JA, et al. The principal eosinophil peroxidase product, HOSCN, is a uniquely potent phagocyte oxidant inducer of endothelial cell tissue factor activity: a potential mechanism for thrombosis in eosinophilic inflammatory states. Blood 2006; 107:558.
36) Moosbauer C, Morgenstern E, Cuvelier SL, et al. Eosinophils are a major intravascular location for tissue factor storage and exposure. Blood 2007; 109:995.
37) Moore PM, Harley JB, Fauci AS. Neurologic dysfunction in the idiopathic hypereosinophilic syndrome. Ann Intern Med 1985; 102:109.
38) Aida L, Parkhutik V, Tembl JI, et al. Embolism and impaired washout: a possible explanation of border zone strokes in hypereosinophilic syndrome. J Neurol Sci 2013; 325:162.
39) Davis SD, Schaller J, Wedgwood RJ. Job`s Syndrome. Recurrent, “cold”, staphylococcal abscesses. Lancet 1966; 1:1013.
40) Hill HR, Ochs HD, Quie PG, et al. Defect in neutrophil granulocyte chemotaxis in Job`s syndrome of recurrent “cold” staphylococcal abscesses. Lancet 1974; 2:617.
41) Van Scoy RE, Hill HR, Ritts RE, Quie PG. Familial neutrophil chemotaxis defect, recurrent bacterial infections, mucocutaneous candidiasis, and hyperimmunoglobulinemia E. Ann Intern Med 1975; 82:766.
42) Grimbacher B, Holland SM, Gallin JI, et al. Hyper-IgE syndrome with recurrent infections–an autosomal dominant multisystem disorder. N Engl J Med 1999; 340:692.
43) Jain N, Cortes J, Quintás-Cardama A, et al. Imatinib has limited therapeutic activity for hypereosinophilic syndrome patients with unknown or negative PDGFRalpha mutation status. Leuk Res 2009; 33:837.
44) Weller PF, Bubley GJ. The idiopathic hypereosinophilic syndrome. Blood 1994; 83:2759.
45) Butterfield JH, Weiler CR. Treatment of hypereosinophilic syndromes–the first 100 years. Semin Hematol 2012; 49:182.
46) Vaklavas C, Tefferi A, Butterfield J, et al. `Idiopathic` eosinophilia with an Occult T-cell clone: prevalence and clinical course. Leuk Res 2007; 31:691.
47) Razaq W, Beautyman E. Successful treatment of refractory idiopathic hypereosinophilic syndrome with etoposide. Am J Ther 2009; 16:68.
48) Klion AD, Bochner BS, Gleich GJ, et al. Approaches to the treatment of hypereosinophilic syndromes: a workshop summary report. J Allergy Clin Immunol 2006; 117:1292.
49) Klion AD, Law MA, Noel P, et al. Safety and efficacy of the monoclonal anti-interleukin-5 antibody SCH55700 in the treatment of patients with hypereosinophilic syndrome. Blood 2004; 103:2939.
50) Plötz SG, Simon HU, Darsow U, et al. Use of an anti-interleukin-5 antibody in the hypereosinophilic syndrome with eosinophilic dermatitis. N Engl J Med 2003; 349:2334.
51) Stein ML, Villanueva JM, Buckmeier BK, et al. Anti-IL-5 (mepolizumab) therapy reduces eosinophil activation ex vivo and increases IL-5 and IL-5 receptor levels. J Allergy Clin Immunol 2008; 121:1473.
52) Verstovsek S, Tefferi A, Kantarjian H, et al. Alemtuzumab therapy for hypereosinophilic syndrome and chronic eosinophilic leukemia. Clin Cancer Res 2009; 15:368.
53) Kalac M, Quintás-Cardama A, Vrhovac R, et al. A critical appraisal of conventional and investigational drug therapy in patients with hypereosinophilic syndrome and clonal eosinophilia. Cancer 2007; 110:955.
54) Esteva-Lorenzo FJ, Meehan KR, Spitzer TR, Mazumder A. Allogeneic bone marrow transplantation in a patient with hypereosinophilic syndrome. Am J Hematol 1996; 51:164.
55) Ueno NT, Anagnostopoulos A, Rondón G, et al. Successful non-myeloablative allogeneic transplantation for treatment of idiopathic hypereosinophilic syndrome. Br J Haematol 2002; 119:131.
56) Halaburda K, Prejzner W, Szatkowski D, et al. Allogeneic bone marrow transplantation for hypereosinophilic syndrome: long-term follow-up with eradication of FIP1L1-PDGFRA fusion transcript. Bone Marrow Transplant 2006; 38:319.
57) Bergua JM, Prieto-Pliego E, Román-Barberá A, et al. Resolution of left and right ventricular thrombosis secondary to hypereosinophilic syndrome (lymphoproliferative variant) with reduced intensity conditioning allogenic stem cell transplantation. Ann Hematol 2008; 87:937.